Atoms or molecules can diffuse rapidly on surfaces covered by other adsorbants
Chemical processes generally involve the transport of atoms and molecules, which on the nanoscale is dominated by diffusion. In interface chemistry, diffusion along surfaces and interfaces is an elementary and often determining step. For example, it plays a central role in crystal growth and dissolution, the deposition of thin films and nanostructures, and the self-assembly of two-dimensional organic layers. It also governs the transport of the reacting species on the surfaces of catalysts and thus the speed by which these species intermix or reach specific sites, where the catalytic reaction occurs. On page 715 of this issue, Henß et al. show that this transport can be rapid even in the presence of a layer of other adsorbants on the surface (1, 2).
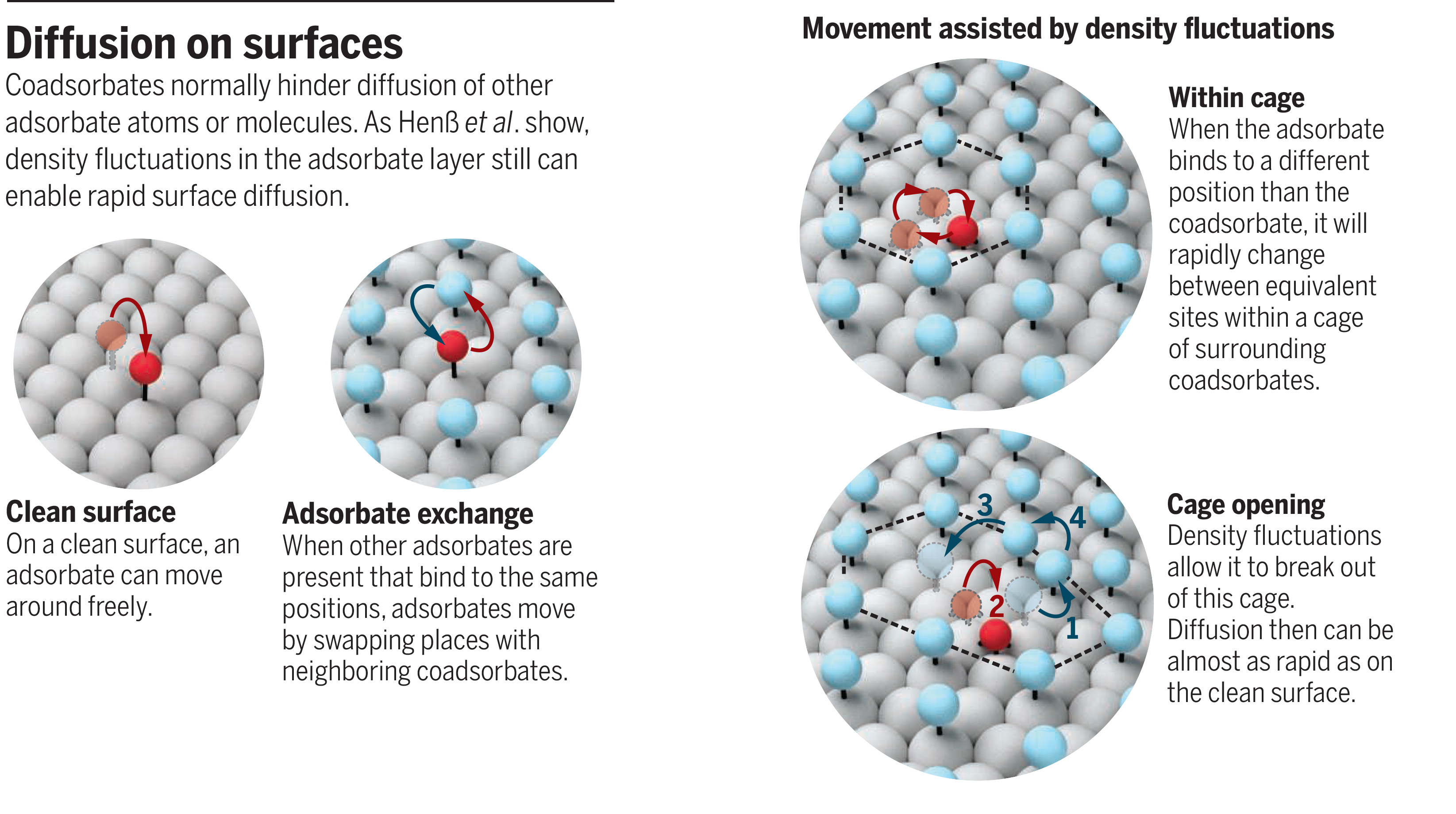
Surface diffusion has been extensively studied for chemically bound atoms and small molecules that cover a small fraction of an otherwise clean solid surface (3). These adsorbates typically move on the surface by jumping between neighboring binding sites (see the figure, top left). During a jump, they must transiently occupy positions in which they are bound less strongly. This leads to an energy barrier, which the adsorbates overcome through thermal excitation. At a given temperature, the rate by which jumps between different positions on the surface lattice occur thus depends on the spatial variation of the binding energy; that is, it is exclusively determined by the interactions of the adsorbate with the atoms of the underlying solid.
This simple picture is, however, usually insufficient to fully describe surface diffusion in the real world. Even if only one species is present on the surface, the adsorbates may encounter other adsorbates during their random walk on the surface and interact with those. In the simplest case, the only effect of this is that positions already occupied by adsorbates are not available to others, a concept called “site blocking.” In reality, however, the adsorbates additionally attract or repulse each other, resulting in modified energy barriers for diffusion in the vicinity of other adsorbates. If the surface fraction covered by adsorbates is high, these effects can lead to changes in the adsorbates mobility by orders of magnitude (4).
Similar effects may be expected for the motion of adsorbates on a surface covered by another atomic or molecular species, which Henß et al. address in their report. The diffusion of adsorbates on surfaces crowded by other coadsorbed species is the normal case for interface processes in ambient or high-pressure gas and liquid phases. However, despite its prevalence in technological systems and natural environments, it has been studied only sparsely and is not well understood. For this reason, the influence of coadsorbates on surface diffusion has mostly been either ignored or treated by simplified concepts such as site blocking.
In the past decade, the complexity of this situation has been revealed in some in-depth studies of adsorbate systems with well-defined composition and geometry (5, 6). These studies combine experimental data on surface diffusion, obtained from video sequences of atomic-resolution scanning tunneling microscopy images, with density functional theory calculations of the pathways by which the adsorbates move between neighboring sites. Hsieh et al. have investigated the diffusion of hydrogen on a chlorine-covered silicon surface and observed a direct exchange of the hydrogen atoms with neighboring chlorine atoms (5). They attributed the surprisingly low energy barrier for this exchange diffusion to the transient formation of a more weakly bound HCl adsorbate. Rahn et al. studied the diffusion of sulfur atoms on copper electrodes that were immersed in aqueous solution and covered by dense halide adlayers (6). They found an opposite dependence of the sulfur diffusion rates on the electrode voltage for sulfur coadsorbed with chloride and bromide, respectively, indicating different diffusion mechanisms for the two coadsorbates.
In both studies, the diffusing adsorbates and the surrounding coadsorbate were bound in an identical geometry to the surface and had the same distances to neighboring adsorbates (see the figure, top right). The diffusion processes can then be conveniently described via motions on one lattice that is common to both species. However, adsorbate and coadsorbate often bind differently to the surface and thus do not occupy identical adsorption sites. How surface diffusion proceeds in this more complex case is the central topic of the report by Henß et al.
As an example, they chose the diffusion of oxygen atoms on a ruthenium surface covered by carbon monoxide, which is an important model system with relevance to the catalytic oxidation of CO. On the hexagonal surface lattice of the basal plane of Ru, oxygen atoms occupy a hollow site, situated between three neighboring Ru atoms, whereas CO is located on top of a Ru surface atom. Because of this mismatch, three symmetrical oxygen-binding sites exist within the cage formed by the surrounding CO (see the figure, bottom). Jumps between the three positions within the cage are not impeded by the CO coadsorbates. They thus occur much more frequently than jumps to the other neighboring binding sites on the Ru surface, which require relocation of a CO molecule. Nevertheless, the latter jumps also occur at a surprisingly high rate—nearly as high as on the CO-free Ru surface.
Henß et al. explain this observation with a mechanism in which first one CO steps aside, moving closer to the neighboring CO adsorbates. This opens a door in the cage by which the oxygen atom can escape to an adjacent site, followed by CO rearrangement to restore the coadsorbate lattice (see the figure, bottom). Density functional calculations show this door opening to be energetically preferred both to a mechanism in which the oxygen atom jumps first and triggers the necessary CO displacement and to a concerted ringlike exchange of O and CO adsorbates.
In this scenario, the coadsorbates do not necessarily represent static obstacles to adsorbate diffusion, as implied in the site blocking concept. Rather, natural dynamic fluctuations in the density of the coadsorbate lattice can enable efficient transport pathways for the embedded diffusing species. One might expect the surrounding coadsorbate layer to nevertheless still lead to a strong reduction in the diffusion rate. The reason why this is not the case in the system studied by Henß et al. is probably that although the CO adlayer is ordered, its density is only 50% of the saturation value. This facilitates the fluctuations required for this mechanism. Future studies should clarify the effect of adlayer density and in-plane order. In addition to the dynamic effects highlighted by Henß et al., coadsorbed layers may also weaken or modify the adsorbate’s binding to the surface and thus lower the energy barriers that result from the adsorbate-surface interaction or even change the diffusion mechanism.
Henß et al.’s findings show that the influence of coadsorbates on adsorbate diffusion are complex and do not necessarily result in a reduction of the surface mobility. This has important consequences for understanding real-world interface processes. In the field of interface reactions, such as in heterogeneous catalysis, high mobility ensures rapid intermixing of the species on the surface. As pointed out by Henß et al., this rapid intermixing is of substantial relevance for macroscopic reaction kinetics. Fast diffusion is also important for mineralization, technological deposition, and organic self-assembly. With better understanding of the way by which coadsorbates control surface transport, these processes may be fine-tuned through the targeted development of suitable additives
REFERENCES
A.-K. Henß et al., Science 363, 715 (2019).
G. Antczak, G. Ehrlich, Surface Diffusion: Metals, Metal Atoms, and Clusters (Cambridge Univ. Press, 2010).
J. V. Barth, Surf. Sci. Rep. 40, 75 (2000).
M.-T. Nguyen, P. N. Phong, J. Phys. Chem. A 121, 5520 (2017).
M.-F. Hsieh, D.-S. Lin, S.-F. Tsay, Phys. Rev. B 80, 045304 (2009).
B. Rahn et al., Angew. Chem. Int. Ed. 57, 6065 (2018).